

PlayMolecule Glimpse: Understanding Protein–Ligand Property Predictions with Interpretable NN
- kübra:)
- Feb 13, 2022
- 4 min read
PlayMolecule Glimpse: Understanding Protein-Ligand Property Predictions with Interpretable Neural Networks
Deep learning has been successfully applied to structure-based protein-ligand affinity prediction, yet the black box nature of these models raises some questions. In a previous study, they presented KDEEP, a convolutional neural network that predicted the binding affinity of a given protein–ligand complex while reaching state-of-the-art performance. However, it was unclear what this model was learning. In this work, they present a new application to visualize the contribution of each input atom to the prediction made by the convolutional neural network, aiding in the interpretability of such predictions. The results suggest that KDEEP is able to learn meaningful chemistry signals from the data, but it has also exposed the inaccuracies of the current model, serving as a guideline for further optimization of their prediction tools.

Machine-learning methods have been widely applied in the field of chemoinformatics, ranging from simple, regressor-based QSAR models to more complex neural networks. These latter methods have been reported to increase performance in some critical tasks for drug discovery, such as toxicity assessment, pharmacokinetics, physicochemical property prediction, and protein–ligand binding affinity prediction.

Main view of the graphical user interface. The protein–ligand complex is displayed, with the attributions of the most contributing voxels superimposed. The attributions for the different channels can be seen individually using the corresponding sliders in the menu on the right, which display isosurfaces at different isovalues. The full protein is shown in a cartoon representation, while residues in the binding site (defined by being within 5 Å to the ligand) are shown in a transparent ball–stick representation (only heavy atoms and polar hydrogens). The all-atom representation of the ligand is shown in a bold ball–stick. The region of space seen by the model (voxelization cube) is delimited by a transparent, gray box.
In a previous work, they developed KDEEP–a 3D convolutional neural network (CNN) that accepts as input a voxelized representation of a protein–ligand complex and outputs a prediction of binding affinity with state-of-the-art accuracy. However, it was unclear whether KDEEP was learning meaningful chemistry or just exploiting shortcuts such as the positive relationship between molecular weight and affinity. Learning these shortcuts instead of the underlying nature of the problem is a topic of concern in the field. It is then comprehensible for many machine learning methods to spark criticism regarding the difficulty to understand the rationale behind their predictions. It has been questioned whether a pharmaceutical company would promote a given molecule into a portfolio based only on an opaque prediction made by a neural network, without any clear explanation to support it. Providing such an explanation would undoubtedly increase the value, trustworthiness, and usability of machine learning models in drug discovery.
Recently, advances in model interpretability, as well as the availability of software libraries such as Captum and Alibi, have allowed researchers to get a first glimpse of what features of the input are more influential toward predictions made by neural networks (i.e., feature attribution assignment). One natural approach to measure this influence is to look at the gradients of the output neuron with respect to the input. In fact, in a CNN trained to discriminate accurate from inaccurate binding poses and to predict binding affinity, visually inspecting these gradients can reveal in which direction the atoms should move to improve the score that the network assigns it, providing some degree of interpretability.

Comparison of computed attributions obtained for a complex of HSP90 with an analogue of benzamide tetrahydro-4H-carbazol-4-one (PDB code: 3D0B) by the three models: Clash detector (1A and 1B), Pose classifier (2A and 2B), and KDEEP (3A and 3B). Pictures on the top row show the attributions for the protein and ligand occupancy channels, in red and blue, respectively. The bottom row focuses on particular interactions. 1B shows a clash between the ligand and the leucine and the attributions for the occupancy channels of protein and ligand (red and blue). 2B and 3B show the hydrogen bond between the benzamide moiety in the ligand and the aspartate (D93) residue in the protein. Attributions for the ligand donor channel are shown in pink, while for the protein acceptor channel, the attributions are shown in blue.
However, backpropagating the prediction relative to the input layer can produce very low gradients in the vicinity of the input vector, a process which is known as gradient saturation. The Integrated Gradients (IG) feature attribution technique helps to mitigate this problem, providing a better measure of how each input feature influences the prediction. Instead of evaluating the gradients at one particular input value (the image in a traditional 2D-CNN), gradients are computed for several variants of that image, ranging from a user-defined baseline (typically, an image with all its pixel-channel values set to zero) to the actual image.
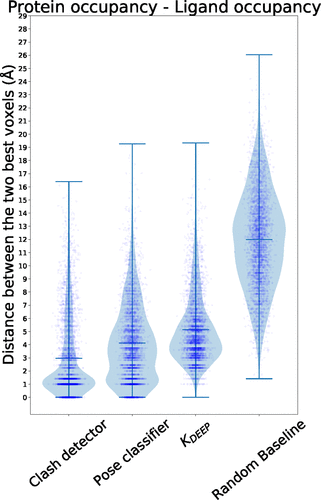
Distance distribution between the two voxels with the highest, absolute value in protein and ligand occupancy channels.
In each variant, the values of all its pixels are multiplied by a scalar α, ranging from the zeroed-out input to the original image. At low values of α, the resulting input vector is far from the usual input space the network has been exposed to during training, circumventing the gradient saturation issue. In this article, they present an application to visualize the contribution of the input features for the prediction of KDEEP and similar CNNs. In addition to describing the methodology used herein, they also showcase several relevant examples of attributions which match with structural biology knowledge. They analyze the prediction of three distinct models: a clash detector, a docking pose classifier, and KDEEP. The clash detector provides a baseline to which they compare the other models and allowed us to validate the implementation of this application. The docking pose classifier and KDEEP models were evaluated to see if CNNs trained to perform chemically relevant tasks were learning meaningful chemistry. The application, called Glimpse, is available to use at https://www.playmolecule.org/Glimpse/.
PlayMolecule Glimpse: Understanding Protein–Ligand Property Predictions with Interpretable Neural Networks
Alejandro Varela-Rial, Iain Maryanow, Maciej Majewski, Stefan Doerr, Nikolai Schapin, José Jiménez-Luna, and Gianni De Fabritiis
Journal of Chemical Information and Modeling 2022 62 (2), 225-231
DOI: 10.1021/acs.jcim.1c0069
Comments